Integration (iRECODE)
We demonstrate RECODE integration for scRNA-seq data produced by 10X Chromium. We use scRNA-seq datasets encompassing three batches and two cell lines (HEK293T and Jurkat). The test data is directly available from GitHub page of JinmiaoChenLab. All data can be downloaded by “git clone https://github.com/JinmiaoChenLab/Batch-effect-removal-benchmarking.git”
We use scanpy to read/write 10X data. Import numpy and scanpy in addlition to screcode.
[3]:
#pip install screcode harmonypy anndata
[1]:
import screcode
import scanpy as sc
import anndata
import numpy as np
import pandas as pd
import sklearn
import warnings
warnings.simplefilter('ignore')
Read in the count matrix into an AnnData object.
[5]:
input_dir = 'Batch-effect-removal-benchmarking/Data/dataset6'
input_filenames = ['b1_exprs','b2_exprs','b3_exprs']
input_filenames_meta = ['b1_celltype','b2_celltype','b3_celltype']
data_input = pd.concat([pd.read_csv('%s/%s.txt.gz' % (input_dir,file),index_col=0,delimiter='\t').T for file in input_filenames])
data_input_meta = pd.DataFrame()
for i in range(len(input_filenames)):
df_ = pd.read_csv('%s/%s.txt.gz' % (input_dir,input_filenames_meta[i]),index_col=0,delimiter='\t')
df_["batch"] = np.full(len(df_.index),input_filenames[i],dtype=object)
data_input_meta = pd.concat([data_input_meta,df_])
adata = anndata.AnnData(X=data_input,obs=data_input_meta)
adata = adata[:,np.sum(adata.X,axis=0)>0]
adata.layers["Raw"] = adata.X
adata
[5]:
AnnData object with n_obs × n_vars = 9531 × 21474
obs: 'CellType', 'batch'
layers: 'Raw'
Apply iRECODE
Apply iRECODE to the count matrix. The anndata or ndarray data formats are available. If your data format is ndarray, prepare meta_data including batch infromation by DataFrame format.
[6]:
recode = screcode.RECODE()
adata = recode.fit_transform(adata)
start RECODE for scRNA-seq data
end RECODE for scRNA-seq
log: {'seq_target': 'RNA', '#significant genes': np.int64(14462), '#non-significant genes': np.int64(7012), '#silent genes': np.int64(0), 'ell': np.int64(154), 'Elapsed time': '0h 0m 30s 429ms', 'solver': 'full'}
[7]:
## Case of ndarray format
## data: single-cell count matrix [ndarray], meta_data: meta data including batch labels [DataFrame]
# recode = screcode.RECODE()
# data_RECODE = recode.fit_transform(data, meta_data=meta_data, batch_key="batch")
With anndata format, outputs of RECODE are included in anndata objects:
denoised matrix -> adata.obsm[‘RECODE’]
noise variance -> adata.var[‘noise_variance_RECODE’]
normalized variance (NVSN variance) -> adata.var[‘normalized_variance_RECODE’]
clasification of genes (significant/non-significant/silent) -> adata.var[‘significance_RECODE’]
Performance check
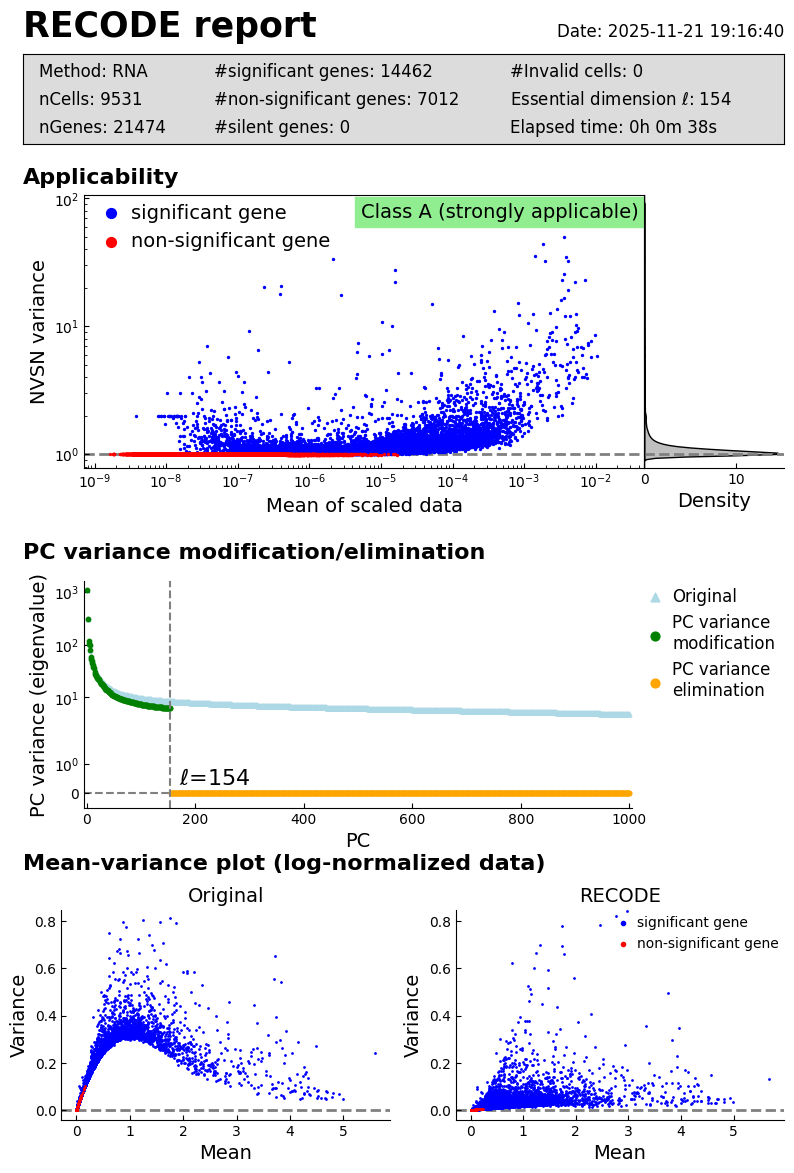
[8]:
recode.report()
Downstream analysis
log normalization
[9]:
target_sum = 1e4
adata.layers["Raw_log"] = np.log(target_sum*adata.layers["Raw"]/np.sum(adata.layers["Raw"],axis=1)[:,np.newaxis]+1)
adata.layers["RECODE_log"] = np.log(target_sum*adata.layers["RECODE"]/np.sum(adata.layers["RECODE"],axis=1)[:,np.newaxis]+1)
PCA
[10]:
n_components = 50
adata.obsm["Raw_PCA"] = sklearn.decomposition.PCA(n_components=n_components).fit_transform(adata.layers["Raw_log"])
adata.obsm["RECODE_PCA"] = sklearn.decomposition.PCA(n_components=n_components).fit_transform(adata.layers["RECODE_log"])
[11]:
import matplotlib
import matplotlib.pyplot as plt
def plot_2d(
adata,
data_lables = ["Raw", "RECODE"],
data_type = "PCA",
color_labels = ['batch','CellType'],
fig_size = 4,
fs_title = 16,
fs_label = 14,
fs_legend = 14,
labels = ["PC1","PC2"],
cmaps = ["tab10","tab10_r"],
vmaxs = [10,10]
):
fig,ax = plt.subplots(2,2,figsize=((len(data_lables)+0.5)*fig_size,len(color_labels)*fig_size),tight_layout=True)
for i in range(len(color_labels)):
colors = np.copy(adata.obs[color_labels[i]].values)
color_set = np.unique(colors)
colors_num = np.copy(colors)
[np.place(colors_num,colors_num==color_set[i],i) for i in range(len(color_set))]
for j in range(len(data_lables)):
ax_ = ax[i,j]
name_ = data_lables[j] + "_" + data_type
plot_data = adata.obsm[name_]
idx_rdm_ = np.random.permutation(plot_data.shape[0])
ax_.scatter(plot_data[idx_rdm_,0],plot_data[idx_rdm_,1],c=colors_num[idx_rdm_],s=1,cmap=cmaps[i],vmax=vmaxs[i],zorder=100)
ax_.tick_params(bottom=False, left=False, right=False, top=False, labelbottom=False, labelleft=False, labelright=False, labeltop=False)
if j==len(data_lables)-1:
for c_ in np.unique(colors):
idx_ = colors == c_
ax_.scatter(plot_data[idx_,0],plot_data[idx_,1],c=colors_num[idx_],s=1,cmap=cmaps[i],vmin=0,vmax=vmaxs[i],label=c_,zorder=0)
ax_.legend(bbox_to_anchor=(1.0,0.5), loc="center left", borderaxespad=0, fontsize=fs_legend,markerscale=10,ncol=1,frameon=False,handletextpad=0.,borderpad=0)
if i == 0:
ax_.set_title(data_lables[j],fontsize=fs_title)
plt.gcf().text(0.435,0.0,labels[0], ha="center",va="center",fontsize=fs_label)
plt.gcf().text(0.0,0.49,labels[1], ha="center",va="center",rotation=90, fontsize=fs_label)
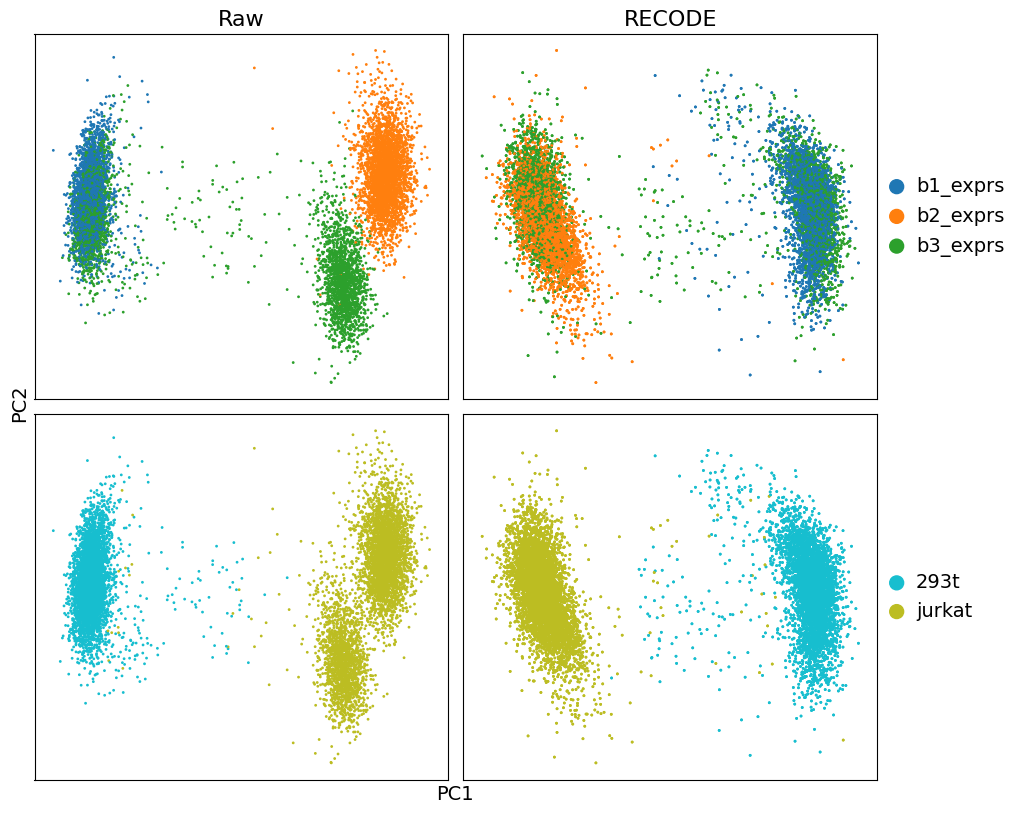
plot_2d(
adata,
data_lables = ["Raw", "RECODE"],
data_type = "PCA",
color_labels = ['batch','CellType'],
labels = ["PC1","PC2"]
)
UMAP
[12]:
import umap
n_components = 2
adata.obsm["Raw_UMAP"] = umap.UMAP(n_components=n_components).fit_transform(adata.obsm["Raw_PCA"])
adata.obsm["RECODE_UMAP"] = umap.UMAP(n_components=n_components).fit_transform(adata.obsm["RECODE_PCA"])
[13]:
plot_2d(
adata,
data_lables = ["Raw", "RECODE"],
data_type = "UMAP",
color_labels = ['batch','CellType'],
labels = ["UMAP1","UMAP2"]
)
[14]:
def plot_gex(
adata,
genes,
data_lables = ["Raw_log", "RECODE_log"],
color_labels = ['batch','CellType'],
fig_size = 4,
fs_title = 16,
fs_label = 14,
fs_legend = 14,
cmaps = ["tab10","tab10_r"],
vmaxs = [10,10]
):
for gene_ in genes:
fig,ax = plt.subplots(2,len(data_lables),figsize=((len(data_lables)+0.5)*fig_size,len(color_labels)*fig_size),tight_layout=True)
for i in range(len(color_labels)):
colors = np.copy(adata.obs[color_labels[i]].values)
color_set = np.unique(colors)
colors_num = np.copy(colors)
[np.place(colors_num,colors_num==color_set[i],i) for i in range(len(color_set))]
for j in range(len(data_lables)):
ax_ = ax[i,j]
name_ = data_lables[j]
plot_data = adata.layers[name_]
idx_x_ = adata.var.index == gene_[0]
idx_y_ = adata.var.index == gene_[1]
idx_rdm_ = np.random.permutation(plot_data.shape[0])
ax_.scatter(plot_data[idx_rdm_][:,idx_x_],plot_data[idx_rdm_][:,idx_y_],c=colors_num[idx_rdm_],s=1,cmap=cmaps[i],vmax=vmaxs[i],zorder=100)
ax_.tick_params(bottom=False, left=False, right=False, top=False, labelbottom=False, labelleft=False, labelright=False, labeltop=False)
if j==len(data_lables)-1:
for c_ in np.unique(colors):
idx_ = colors == c_
ax_.scatter(plot_data[idx_][:,idx_x_],plot_data[idx_][:,idx_y_],c=colors_num[idx_],s=1,cmap=cmaps[i],vmin=0,vmax=vmaxs[i],label=c_,zorder=0)
ax_.legend(bbox_to_anchor=(1.0,0.5), loc="center left", borderaxespad=0, fontsize=fs_legend,markerscale=10,ncol=1,frameon=False,handletextpad=0.,borderpad=0)
if i == 0:
ax_.set_title(data_lables[j],fontsize=fs_title)
ax_.grid(ls="--",alpha=0.5,zorder=0)
plt.gcf().text(0.435,0.0,"$\it{%s}$" % gene_[0], ha="center", va="center", fontsize=fs_label)
plt.gcf().text(0.0,0.490,"$\it{%s}$" % gene_[1], ha="center", va="center", fontsize=fs_label,rotation=90)
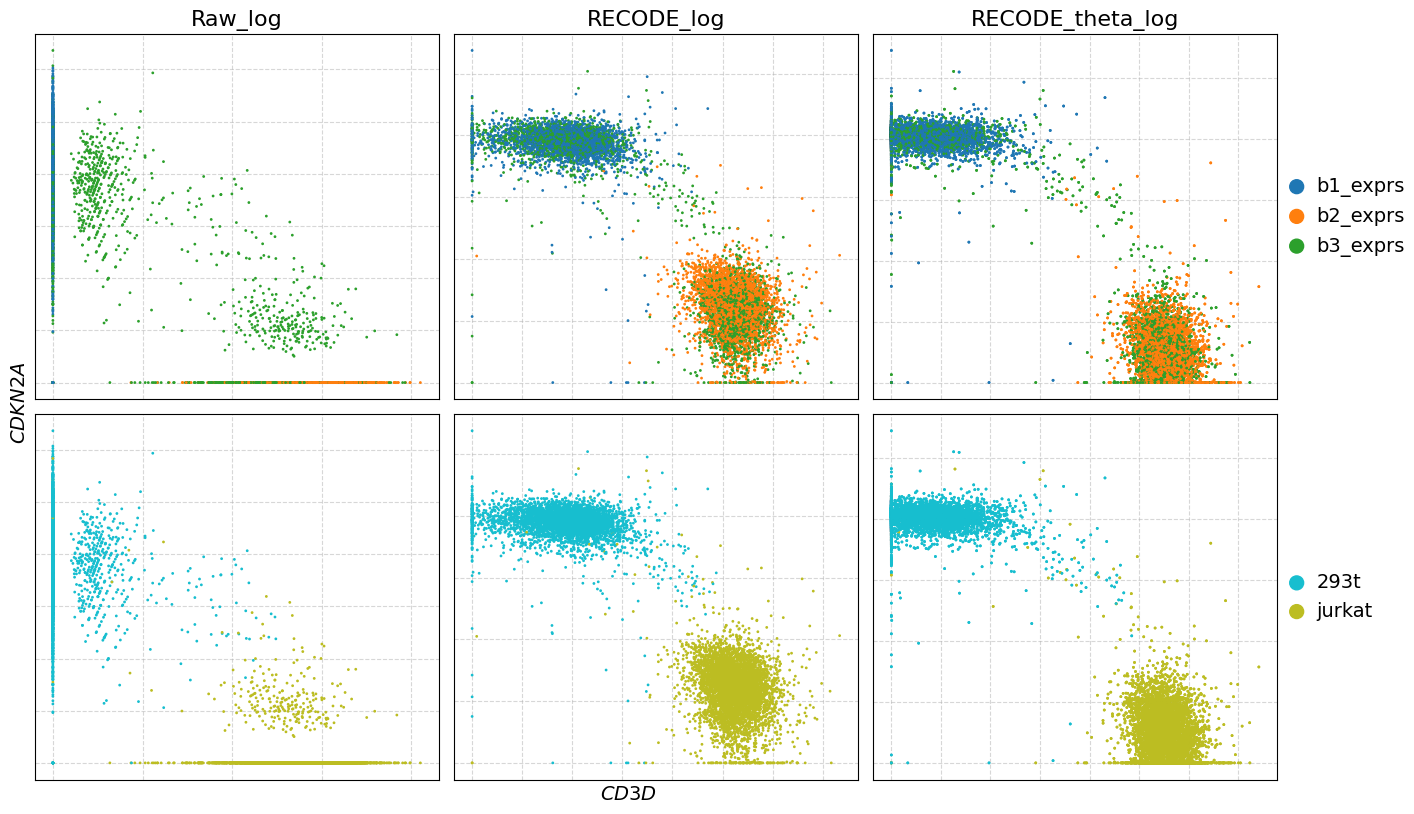
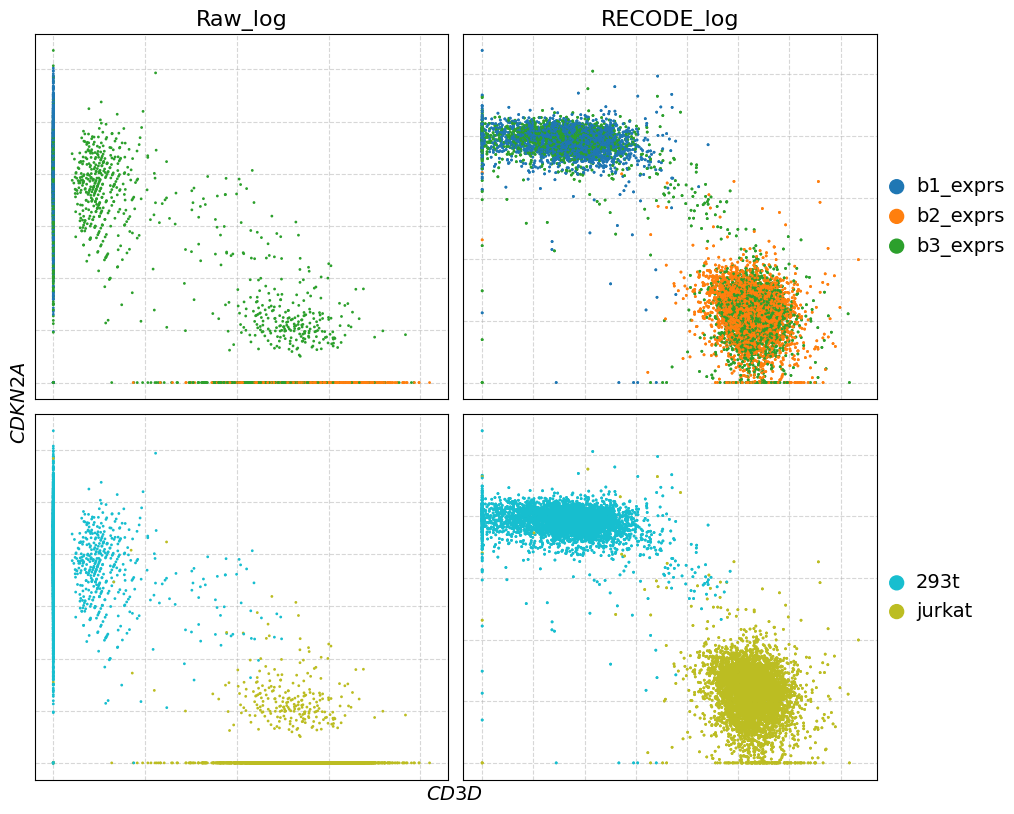
genes = [["CD3D", "CDKN2A"]]
plot_gex(adata,genes)
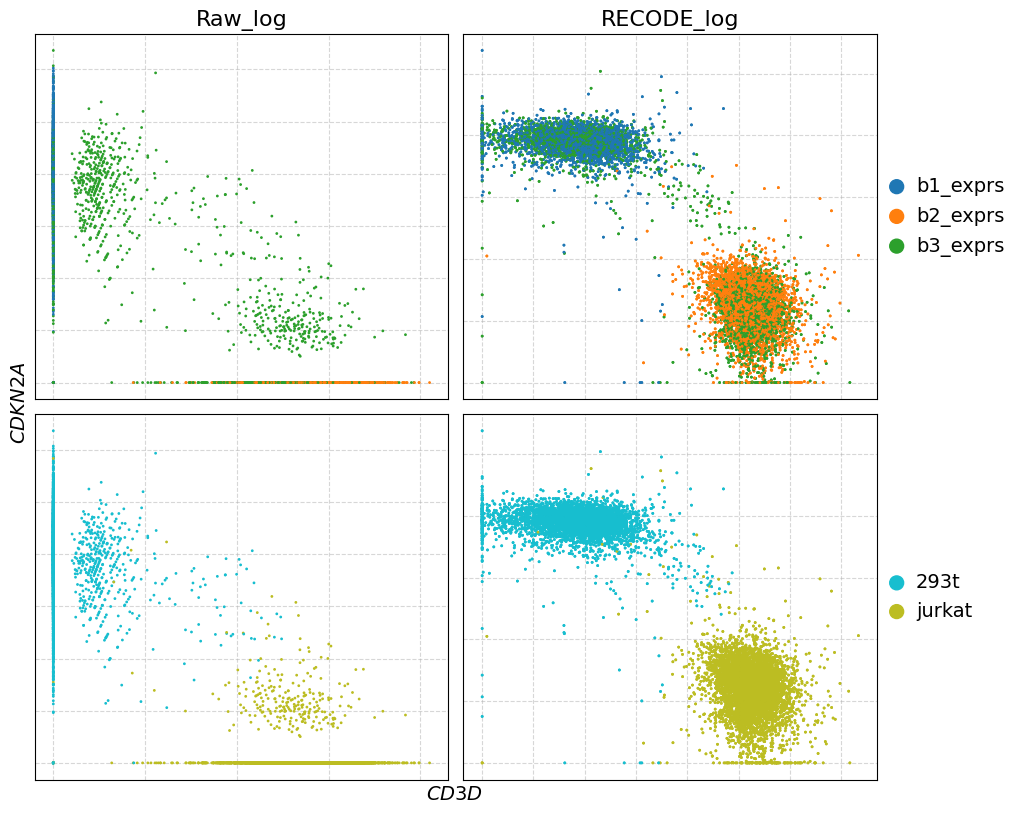
Adjust parameters of batch correction
[15]:
recode = screcode.RECODE(RECODE_key="RECODE_theta")
adata = recode.fit_transform(adata, integration_method_params = {"theta":0.1})
---------------------------------------------------------------------------
TypeError Traceback (most recent call last)
Cell In[15], line 2
1 recode = screcode.RECODE(RECODE_key="RECODE_theta")
----> 2 adata = recode.fit_transform(adata, integration_method_params = {"theta":0.1})
TypeError: RECODE.fit_transform() got an unexpected keyword argument 'integration_method_params'
[ ]:
adata = recode.lognormalize(adata, key="RECODE_theta")
Normalized data are stored as "RECODE_theta_norm" and "RECODE_theta_log" in adata.layers
[ ]:
genes = [["CD3D", "CDKN2A"]]
plot_gex(adata,genes,data_lables = ["Raw_log", "RECODE_log", "RECODE_theta_log"])