Multiome (scRNA-seq + scATAC-seq data)
We show an example of scRNA-seq data produced by 10X Chromium. We are using scATAC-seq data PBMC from a Healthy Donor - No Cell Sorting (3k) (3,009 cells and 117,757 features) from 10X Genomics Datasets. The test data is directly available from Peak by cell matrix HDF5 (filtered) in here (registration required).
We use scanpy to read/write 10X data. Import numpy and scanpy in addlition to screcode.
[15]:
import screcode
import numpy as np
import scanpy as sc
import warnings
warnings.simplefilter('ignore')
Read in the count matrix into an AnnData object.
[16]:
input_filename = 'data/pbmc_unsorted_3k_filtered_feature_bc_matrix.h5'
adata = sc.read_10x_h5(input_filename,gex_only=False)
adata.var_names_make_unique()
adata.layers["Raw"] = adata.X.toarray()
adata
[16]:
AnnData object with n_obs × n_vars = 3009 × 117757
var: 'gene_ids', 'feature_types', 'genome', 'interval'
layers: 'Raw'
[17]:
np.unique(adata.var['feature_types'].values,return_counts=True)
[17]:
(array(['Gene Expression', 'Peaks'], dtype=object), array([36601, 81156]))
Apply RECODE
Apply RECODE to the count matrix. Only anndata data format is available for multiome data.
[18]:
recode = screcode.RECODE(seq_target='Multiome')
adata = recode.fit_transform(adata)
start RECODE for Multiome data
end RECODE for scMultiome-seq
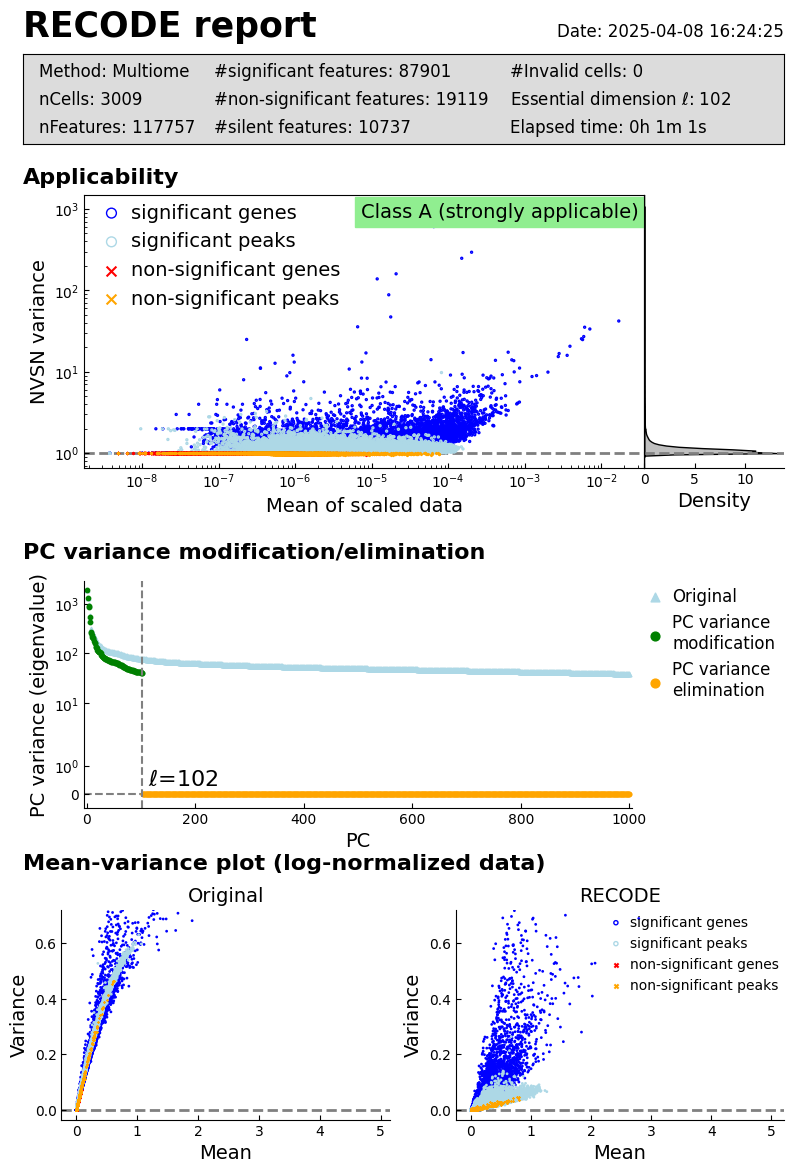
log: {'seq_target': 'Multiome', '#significant features': 87901, '#non-significant features': 19119, '#silent features': 10737, 'ell': 102, 'Elapsed time': '0h 1m 21s 362ms', 'solver': 'full'}
With anndata format, outputs of RECODE are included in anndata objects:
denoised matrix -> adata.obsm[‘RECODE’]
noise variance -> adata.var[‘noise_variance_RECODE’]
normalized variance (NVSN variance) -> adata.var[‘normalized_variance_RECODE’]
clasification of genes (significant/non-significant/silent) -> adata.var[‘significance_RECODE’]
[19]:
adata
[19]:
AnnData object with n_obs × n_vars = 3009 × 117757
var: 'gene_ids', 'feature_types', 'genome', 'interval', 'noise_variance', 'normalized_variance', 'significance_RECODE'
uns: 'RECODE_essential'
layers: 'Raw', 'RECODE', 'RECODE_NVSN'
Performance verification
Show report:
[20]:
recode.report()
[21]:
target_sum = 1e6
adata.X = adata.layers["RECODE"]
adata = recode.lognormalize(adata,target_sum=target_sum)
sc.pp.normalize_total(adata, target_sum=target_sum)
sc.pp.log1p(adata)
Normalized data are stored in "RECODE_norm" and "RECODE_log"
[22]:
adata.layers["Raw_log"] = np.log(target_sum*adata.layers["Raw"]/np.sum(adata.layers["Raw"],axis=1)[:,np.newaxis]+1)
[23]:
import sklearn.decomposition
n_pcs = 50
adata.obsm["X_pca"] = sklearn.decomposition.PCA(n_components=n_pcs).fit_transform(adata.layers["RECODE_log"])
[24]:
sc.tl.pca(adata, svd_solver='arpack')
[25]:
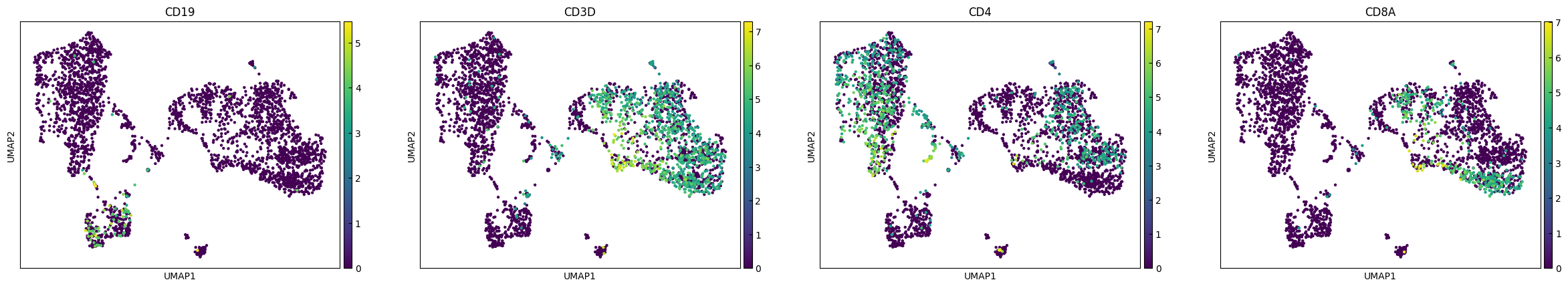
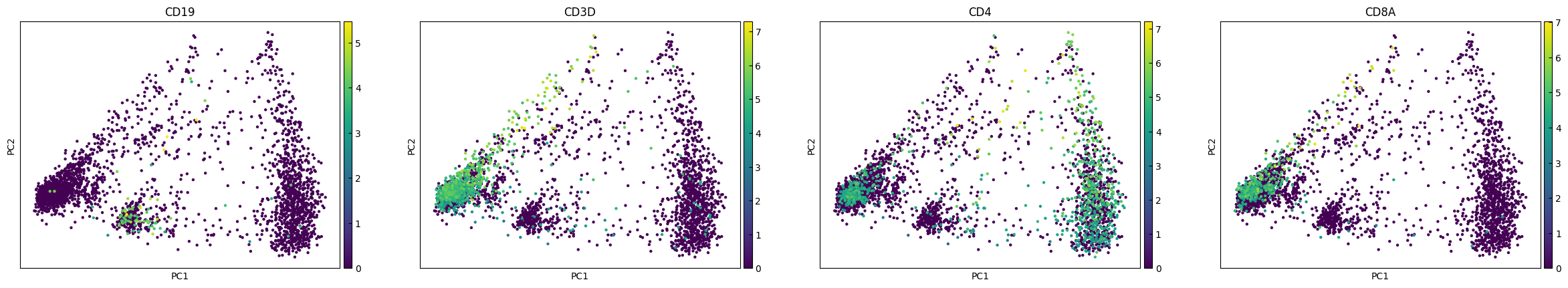
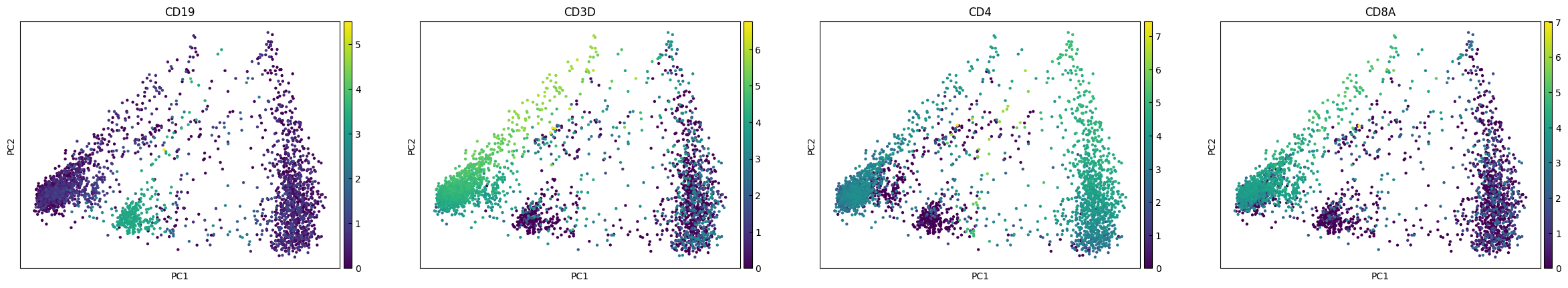
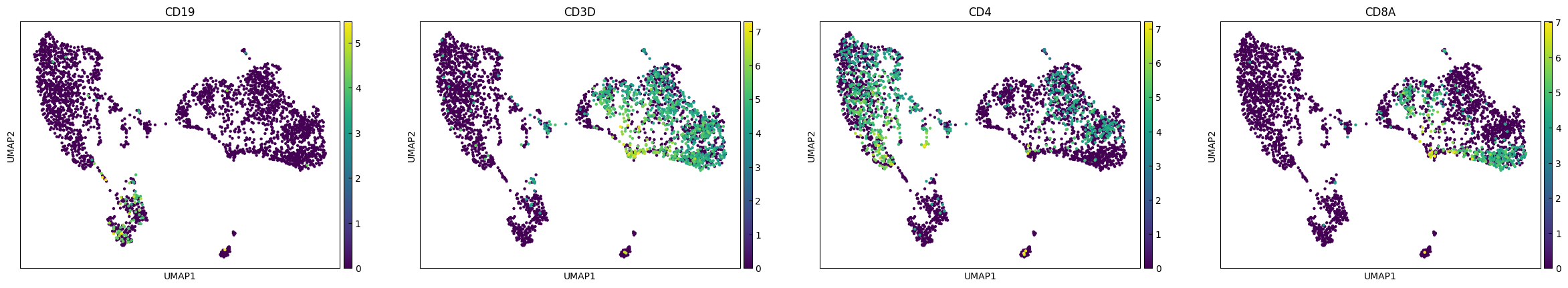
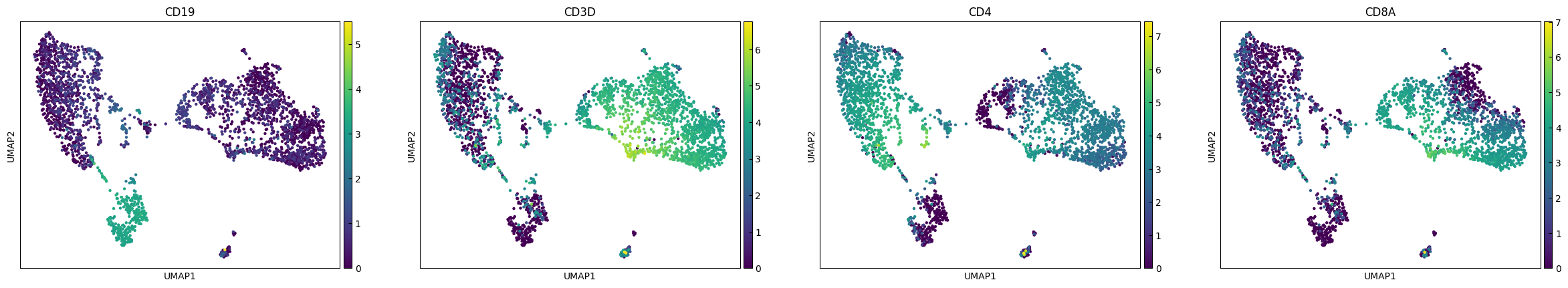
plot_genes = ['CD19','CD3D','CD4','CD8A']
sc.pl.pca(adata, color=plot_genes,layer="Raw_log")
sc.pl.pca(adata, color=plot_genes,layer="RECODE_log")
[26]:
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=0)
sc.tl.umap(adata)
[27]:
sc.pl.umap(adata, color=plot_genes,layer="Raw_log")
sc.pl.umap(adata, color=plot_genes,layer="RECODE_log")
[28]:
sc.tl.leiden(adata,resolution=1)
sc.pl.umap(adata, color=['leiden'])